
In the medical device industry, quality management and regulatory compliance are of paramount importance. Two significant regulations are the European Union’s Medical Device Regulation (MDR 2017/745) and the International Standard Organization’s ISO 13485. Understanding these regulations is crucial for medical device manufacturers, as they define the requirements for a robust Quality Management System (QMS).
MDR 2017/745, implemented by the European Union, is a regulation concerning the manufacturing and distribution of medical devices in the EU market. It emphasizes the need for greater transparency, improved safety, and increased traceability of devices. On the other hand, ISO 13485 prescribes the requirements for a comprehensive QMS for medical devices and holds international recognition.
To ensure compliance and sustainability, medical device manufacturers should familiarize themselves with both MDR 2017/745 and ISO 13485. This understanding will enhance their ability to meet the stringent requirements of both regulatory bodies, providing a competitive edge in the global market.
Understanding ISO 13485: Quality Management System for Medical Devices
ISO 13485 defines the requirements for developing a QMS specifically for medical devices and holds global recognition. The core purpose of this standard is to facilitate harmonized medical device quality management requirements. ISO 13485 places a stronger emphasis on risk management and risk-based decision-making processes, making it significantly different from other QMS standards.
An effective ISO 13485 quality management system ensures that medical devices meet customer expectations and comply with the applicable regulatory requirements. This standard focuses on process control, customer satisfaction, product traceability and recall systems, and maintaining effective corrective and preventive action systems.
Adherence to ISO 13485 demonstrates a commitment to the safety and quality of medical devices, which can strengthen a company’s reputation in the marketplace. However, understanding the requirements of ISO 13485 and implementing a QMS based on this standard can be a challenging task. It entails developing procedures that ensure consistent and compliant processes, which require significant time and effort.
Key Differences between MDR 2017/745 and ISO 13485
While both MDR 2017/745 and ISO 13485 aim to ensure the safety and efficacy of medical devices, there are several key differences. Understanding these differences can help manufacturers navigate these requirements more effectively.
MDR 2017/745, unlike ISO 13485, is a legislative act, which means it’s legally binding. While ISO 13485 focuses on the effectiveness of a QMS. That contains the required processes and procedures and no product-specific requirements. MDR 2017/745 focuses more on the medical device itself with detailed requirements, including its design, development, production, and post-market surveillance.
But are there any additional QMS requirements from the MDR?
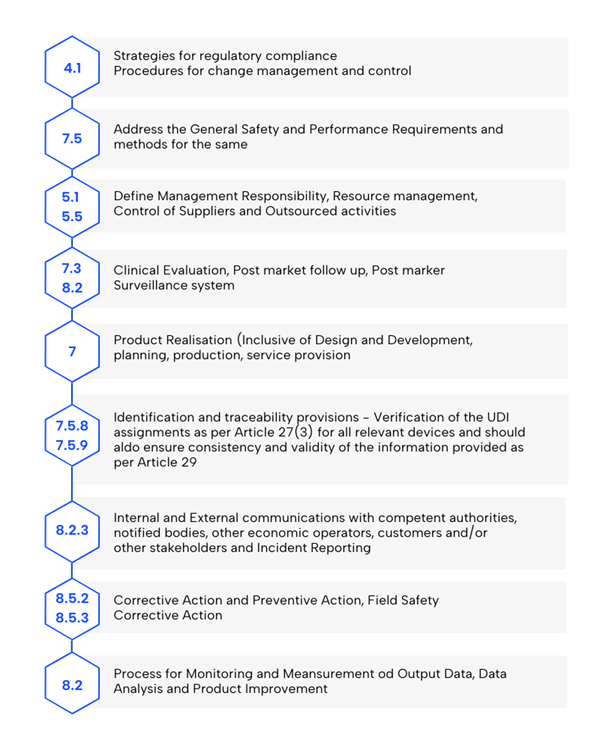
Yes, there are some additions. Find below an overview of key topics to be addressed in your quality management system.
- Monitoring and Data Analysis: Establish processes for measuring outputs, analyzing data, and improving products, with a focus on statistical methods.
- Regulatory Compliance Strategy: Must include conformity assessment procedures and change management.
- General Safety and Performance Requirements (GSPR): Consider Annex I requirements and methods to meet them, ensuring compliance with the MDR.
- Product Realization: Include planning, design, development, production, and service provision, with GSPRs as part of the design requirements.
- Person Responsible for Regulatory Compliance: Define the role as required by Article 15 of the MDR.
- Resource Management: Implement methodologies for selecting and controlling suppliers and subcontractors.
- Risk Management: Establish processes per Section 3, Annex I of the MDR, with EN ISO 14971 filling this regulatory gap.
- Clinical Evaluation and Post-Market Clinical Follow-up (PMCF): Include data as per Article 61 and Annex XIV.
- UDI Verification: Ensure proper assignment for relevant devices and validate data consistency (Articles 27(3) and 29).
- Post-Market Surveillance: Establish and maintain a system as required by Article 83.
- Communication Protocols: Define interactions with authorities, notified bodies, economic operators, customers, and stakeholders.
- Serious Event and Corrective Action Reporting: Include methodologies for vigilance and field safety corrective actions (Article 87).
- Corrective Action Management: Implement and verify corrective actions to ensure effectiveness.
Which parts of ISO 1385 are mainly affected?
Another important difference is that MDR 2017/745 applies exclusively to the European market, while ISO 13485 is recognized globally.
This means a QMS certified to ISO 13485 can be used almost worldwide, whereas compliance with MDR 2017/745 is necessary for accessing the European market.
Despite these differences, both MDR 2017/745 and ISO 13485 have a shared goal: to ensure the safety and performance of medical devices. They both emphasize the importance of a robust QMS, risk management, and continuous improvement.
How will a QMS be assessed under the EU MDR?
The quality management system of a manufacturer will be evaluated as part of the EU MDR Annex IX to XI- conformity assessment procedures. A Notified Body will conduct a conformity evaluation through an audit for all device classes except Class I. That means an existing ISO 13485 certification isn’t sufficient to claim compliance with MDR QMS requirements.
Transitioning from ISO 13485 to MDR 2017/745: A Guide
Transitioning from ISO 13485 to MDR 2017/745 may seem complex, but with the right approach, it can be a smooth process. The first step is to understand the differences between the two regulations and identify the additional requirements posed by MDR 2017/745.
Next, conduct a gap analysis to identify any areas where your current QMS does not meet MDR 2017/745 requirements. This will involve reviewing procedures, documentation, and practices related to all stages of the medical device lifecycle.
Following the gap analysis, develop and implement a plan to address the identified gaps. This may involve updating procedures, enhancing documentation, or strengthening post-market surveillance activities. Be prepared for this to be a time-consuming process, requiring the involvement of various stakeholders.
Finally, ensure that all changes are effectively communicated throughout the organization and that adequate training is provided. This will ensure that everyone understands the new requirements and can contribute to maintaining compliance.
Navigating MDR 2017/745 and ISO 13485 for Quality Management Compliance
Navigating the world of medical device regulations, particularly MDR 2017/745 and ISO 13485, can be complex.
However, understanding these regulations and their differences can help manufacturers develop robust quality management systems and maintain compliance.
Even if the ISO 13485 isn’t mandatory, as the previous chapters demonstrated, it is a strong foundation that will ease the implementation process significantly. Also, the strong focus on product compliance that needs to be reflected in the quality management system needs special attention.
Remember, the goal of both MDR 2017/745 and ISO 13485 is to ensure the safety and efficacy of medical devices. By focusing on this common goal, manufacturers can effectively navigate these regulations and succeed in the global market.
Transitioning from one system to another, or managing compliance with both, can be a significant undertaking. However, with careful planning, clear communication, and ongoing commitment, it is an achievable goal.
If you need support in understanding MDR 2017/745, ISO 13485, or navigating the transition between these regulatory frameworks, don’t hesitate: Talk to our experts!


