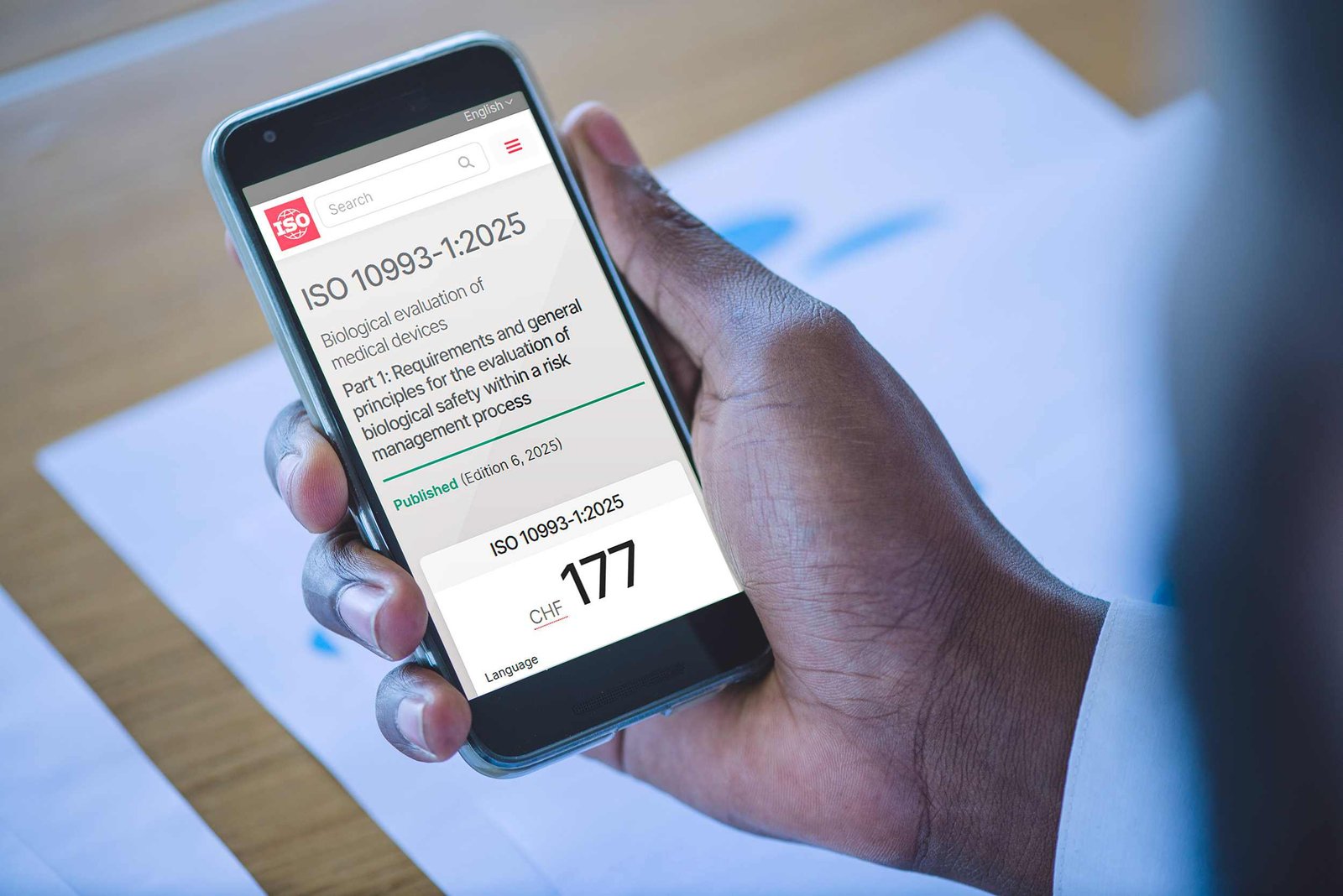
ISO 10993-1 is the international standard that defines the general principles and requirements for the biological evaluation of medical devices according to their categorization.
The ISO 10993 series was developed with a clear purpose: to protect patients from potential biological risks associated with medical devices and the materials from which they are made.
Specifically, ISO 10993-1:2025 is a fundamental pillar for ensuring the safety of medical devices that come into contact with the human body.
Additionally, the standard categorizes devices based on the nature and duration of body contact, allowing for a more precise assessment of associated risks.
For manufacturers, meeting the requirements of this standard may NOT be optional, depending on the device’s categorization. It is a critical requirement for identifying biological risks — and essential for obtaining regulatory approval and marketing medical devices.
We are now at an important moment: the revised ISO 10993-1 for biocompatibility has been published.
In practice, this update brings significant changes that will directly affect how biocompatibility assessments are conducted.
In fact, since the 2018 edition, the industry has increasingly recognized that biological evaluation is not a simple test checklist, but a structured, logic-based process.
Do you want to know what changes with the updated standard and how it impacts medical device manufacturers? Keep reading.
What Is ISO 10993 and Why Is It Important?
ISO 10993-1:2025 is part of an international series of standards that define criteria for the biological evaluation of medical devices, based on the type and duration of contact with the human body.
The standard defines the requirements necessary to determine a device’s potential biological risk by evaluating its biocompatibility — that is, its acceptability for use within the human body.
ISO 10993 Biocompatibility
But how is ISO 10993-1:2025 related to biocompatibility?
The primary purpose of ISO 10993 is to protect humans from potential biological risks associated with the use of medical devices.
As such, Part 1 specifies the general principles that guide biological evaluation within a structured risk-management process.
Furthermore, it establishes a general categorization based on the nature and duration of body contact, making it easier to identify the requirements applicable to each product type.
In this context, the standard favors the use of physicochemical characterization and in vitro methods whenever they provide information as relevant as those obtained from in vivo models.
It is important to note that the Biological Evaluation Report (BER) is not merely a “checklist” of tests but the outcome of a structured plan (BEP) involving:
- Risk analysis: identifying and categorizing the medical device, including its configuration, composition, physical characteristics, and type of body contact during use. Identifying biological hazards, hazardous situations, and biological effects.
- Risk assessment: systematically using available information to identify biological hazards and estimate biological risks. If existing data are insufficient, additional physical, chemical, or biological testing may be required.
- Residual risk evaluation: assessing the biological risk that remains after risk-control measures are implemented.
Main Changes in the New ISO 10993-1 Biocompatibility Revision
ISO 10993-1 has undergone a significant update.
These changes impact how manufacturers and professionals perform biological evaluations of medical devices.
But what exactly changed?
Here are some of the most important updates:
Updated Device Categorization by Contact Type
The new version introduces a revised categorization for device contact types. Devices may now fall under:
- Medical device with no body contact
- Medical device in contact with intact skin
- Medical device in contact with intact mucous membranes
- Medical device in contact with breached or compromised surfaces (skin or mucosa) or internal tissues (excluding circulating blood)
- Medical device in contact with circulating blood
Additionally, when categorizing a device by the duration of contact, the total exposure time must be calculated and classified as Daily Contact or Intermittent Contact.
Finally — and importantly — bioaccumulation must be assessed for all device constituents, regardless of the duration category.
Updated Toxicological Outcomes by Category
The term “biological endpoint” has been replaced by “biological effect”, reflecting an evaluation more focused on potential impacts.
The biological effects table has been reorganized into multiple tables that correspond to each contact categorization.
Stronger Emphasis on Alternative Methods and Animal Welfare
The revision reinforces the 3Rs principle (replace, reduce, refine), restricting in vivo tests to only those strictly necessary.
This shift reflects a growing commitment to ethical practices in biological evaluation, prioritizing alternatives to animal testing whenever feasible.
Integration With the Product Life Cycle
Another major change is the reorganization of the biological evaluation process to align with the risk-management structure defined in ISO 14971.
This integration helps assess biological safety from design through post-market use, considering changes during transport, storage, and use. Thus, the entire device life cycle must be evaluated.
For reusable devices, this includes considering all reprocessing cycles.
More Rigorous Documentation Requirements
Finally, the revision strengthens the requirements for the Biological Evaluation Plan (BEP), which must include or reference criteria for the acceptability of specific biological risks.
Tip: Sobel has prepared a complete guide showing the before-and-after of the ISO 10993-1 Biocompatibility revision!
Just click here to download the material!
What Changes for Medical Device Manufacturers?
With the update to ISO 10993-1, manufacturers and medical device professionals must adapt to a more structured approach that ensures their products remain compliant with the updated requirements for determining biological risk.
Are you part of this scenario? If so, pay close attention to the following points.
Need for Gap Analysis
Gap analysis, already referenced in the previous version of ISO 10993-1, becomes even more prominent in the updated standard as part of the biological evaluation process.
You need to create and maintain this documentation alongside the other reports (BEP and BER) and review it whenever necessary.
For companies with devices previously approved under the 2018 edition of the standard, performing a gap analysis is essential. In these cases, the process helps identify differences between existing documentation and the new requirements.
It’s worth noting that Sobel has highly qualified professionals ready to assist you with this analysis. Send us a message to receive a personalized proposal and prepare for all changes.
Adaptation of Processes and Technical Documentation
Manufacturers will need to determine whether their Biological Evaluation Reports must be updated, which may also require revisions to other documents such as the Risk Management File and Instructions for Use.
Additionally, biological evaluation documentation must include detailed information about the medical device, state of the art, methodological approach used in the biological safety assessment, contact type and exposure duration categorization, justification for performing or waiving tests, risk analysis, and more.
For devices containing nanomaterials, for example, manufacturers must prepare specialized biocompatibility assessment reports.
Importance of Qualified Professionals
Properly trained professionals must conduct the biological evaluation of medical devices.
Ideally, the team responsible for compiling and/or reviewing the updated biological evaluation documentation (Gap Analysis, BEP, and BER) should be qualified, experienced, and well-informed.
This requirement reflects the acknowledgment that biological evaluation demands specialized expertise in ISO 10993 Biocompatibility and toxicology — going far beyond a basic understanding of regulatory requirements.
How to Prepare for the New ISO 10993-1
Will you need to comply with the updated ISO 10993-1 Biocompatibility requirements? Proper preparation requires specific and well-planned actions.

Below are the three essential steps:
- Monitor updates from regulatory authorities: Keep track of communications and updates issued by agencies in your device’s target markets to stay aligned with evolving requirements.
- Review materials and processes used: Choosing appropriate raw materials is a crucial factor. It is recommended to prioritize materials with well-established technical standards and proven historical use.
During this review, consider how processing steps, additives, cleaning agents, and sterilization methods may affect the device’s final composition. - Adopt a proactive, risk-based approach: A proactive approach includes conducting a gap analysis comparing existing documentation to the new version of the standard.
Importance for Patient Safety and Regulatory Compliance
The update to ISO 10993-1 marks a significant milestone for the medical device industry.
ISO 10993 Biocompatibility sets essential parameters to ensure the biological safety of products that come into contact with the human body.
By ensuring material biocompatibility, manufacturers minimize the risk of adverse biological effects such as irritation, sensitization, or more severe outcomes like genotoxicity.
For medical device manufacturers, this represents more than a structural change to the testing framework — it is a regulatory obligation. Without biocompatibility approval, your products may not reach the market.
In addition to the tips shared earlier — including the downloadable PDF — you can count on the Sobel team to guide you throughout this entire journey.
With qualified specialists, we support everything from gap analysis of your documentation to producing BEPs and BERs aligned with the updated ISO 10993 Biocompatibility requirements.
This way, you can feel confident that you are fully prepared for all the changes ahead.


