
No setor de dispositivos médicos, o gerenciamento da qualidade e a conformidade regulamentar são de suma importância. Dois regulamentos importantes são o Regulamento de Dispositivos Médicos da União Europeia (MDR 2017/745) e a ISO 13485 da Organização Internacional de Normalização. Entender essas regulamentações é fundamental para os fabricantes de dispositivos médicos, pois elas definem os requisitos para um Sistema de Gerenciamento de Qualidade (SGQ) robusto.
O MDR 2017/745, implementado pela União Europeia, é um regulamento referente à fabricação e distribuição de dispositivos médicos no mercado da UE. Ele enfatiza a necessidade de maior transparência, segurança aprimorada e maior rastreabilidade dos dispositivos. Por outro lado, a ISO 13485 prescreve os requisitos para um SGQ abrangente para dispositivos médicos e tem reconhecimento internacional.
Para garantir a conformidade e a sustentabilidade, os fabricantes de dispositivos médicos devem se familiarizar tanto com o MDR 2017/745 quanto com a ISO 13485. Esse entendimento aumentará sua capacidade de atender aos rigorosos requisitos de ambos os órgãos reguladores, proporcionando uma vantagem competitiva no mercado global.
Entendendo a ISO 13485: Sistema de Gestão da Qualidade para Dispositivos Médicos
A ISO 13485 define os requisitos para o desenvolvimento de um SGQ especificamente para dispositivos médicos e tem reconhecimento global. O principal objetivo dessa norma é facilitar a harmonização dos requisitos de gerenciamento da qualidade de produtos médicos. A ISO 13485 enfatiza mais o gerenciamento de riscos e os processos de tomada de decisão baseados em riscos, o que a torna significativamente diferente de outros padrões de QMS.
Um sistema de gerenciamento de qualidade ISO 13485 eficaz garante que os dispositivos médicos atendam às expectativas dos clientes e cumpram os requisitos regulamentares aplicáveis. Essa norma se concentra no controle de processos, na satisfação do cliente, na rastreabilidade do produto e nos sistemas de recall, além de manter sistemas eficazes de ações corretivas e preventivas.
A adesão à ISO 13485 demonstra um compromisso com a segurança e a qualidade dos dispositivos médicos, o que pode fortalecer a reputação de uma empresa no mercado. Entretanto, compreender os requisitos da ISO 13485 e implementar um SGQ com base nessa norma pode ser uma tarefa desafiadora. Isso implica o desenvolvimento de procedimentos que garantam processos consistentes e em conformidade, o que exige tempo e esforço significativos.
Principais diferenças entre o MDR 2017/745 e a ISO 13485
Embora tanto o MDR 2017/745 quanto a ISO 13485 tenham como objetivo garantir a segurança e a eficácia dos dispositivos médicos, há várias diferenças importantes. Entender essas diferenças pode ajudar os fabricantes a navegar por esses requisitos de forma mais eficaz.
O MDR 2017/745, diferentemente da ISO 13485, é um ato legislativo, o que significa que é juridicamente vinculativo. Enquanto a ISO 13485 concentra-se na eficácia de um SGQ. Contém os processos e procedimentos necessários e nenhum requisito específico do produto. O MDR 2017/745 se concentra mais no dispositivo médico em si, com requisitos detalhados, incluindo seu projeto, desenvolvimento, produção e vigilância pós-comercialização.
Mas há algum requisito adicional de QMS do MDR?
Sim, há algumas adições. Veja abaixo uma visão geral dos principais tópicos a serem abordados em seu sistema de gerenciamento de qualidade.
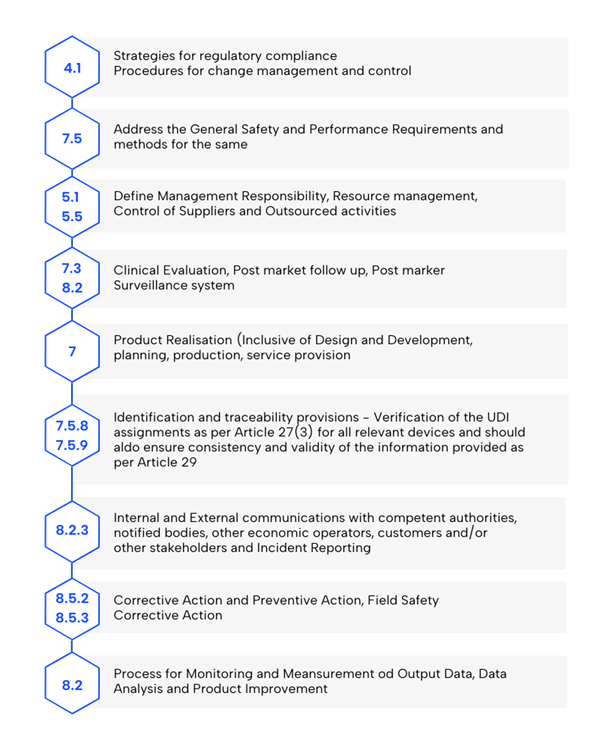
- Monitoramento e análise de dados: Estabelecer processos para medir os resultados, analisar os dados e aprimorar os produtos, com foco em métodos estatísticos.
- Estratégia de conformidade regulatória: Deve incluir procedimentos de avaliação de conformidade e gerenciamento de mudanças.
- Requisitos gerais de segurança e desempenho (GSPR): Considerar os requisitos do Anexo I e os métodos para atendê-los, garantindo a conformidade com o MDR.
- Realização do produto: Incluir planejamento, projeto, desenvolvimento, produção e prestação de serviços, com GSPRs como parte dos requisitos de projeto.
- Pessoa responsável pela conformidade regulatória: Definir a função conforme exigido pelo Artigo 15 do MDR.
- Gerenciamento de recursos: Implementar metodologias para selecionar e controlar fornecedores e subcontratados.
- Gerenciamento de riscos: Estabeleça processos de acordo com a Seção 3, Anexo I do MDR, com a EN ISO 14971 preenchendo essa lacuna regulamentar.
- Avaliação clínica e acompanhamento clínico pós-comercialização (PMCF): Incluir dados de acordo com o Artigo 61 e o Anexo XIV.
- Verificação de UDI: Garantir a atribuição adequada dos dispositivos relevantes e validar a consistência dos dados (artigos 27(3) e 29).
- Vigilância pós-comercialização: Estabelecer e manter um sistema conforme exigido pelo Artigo 83.
- Protocolos de comunicação: Definir interações com autoridades, órgãos notificados, operadores econômicos, clientes e partes interessadas.
- Relatórios de eventos graves e ações corretivas: Incluir metodologias para ações corretivas de vigilância e segurança de campo (Artigo 87).
- Gerenciamento de ações corretivas: Implementar e verificar ações corretivas para garantir a eficácia.
Quais partes da ISO 1385 são mais afetadas?
Outra diferença importante é que a MDR 2017/745 se aplica exclusivamente ao mercado europeu, enquanto a ISO 13485 é reconhecida globalmente.
Isso significa que um QMS certificado pela ISO 13485 pode ser usado em quase todo o mundo, enquanto a conformidade com a MDR 2017/745 é necessária para acessar o mercado europeu.
Apesar dessas diferenças, tanto o MDR 2017/745 quanto a ISO 13485 têm um objetivo em comum: garantir a segurança e o desempenho dos dispositivos médicos. Ambos enfatizam a importância de um SGQ robusto, do gerenciamento de riscos e da melhoria contínua.
Como um QMS será avaliado de acordo com o MDR da UE?
O sistema de gerenciamento de qualidade de um fabricante será avaliado como parte dos procedimentos de avaliação de conformidade dos Anexos IX a XI do MDR da UE. Um Órgão Notificado conduzirá uma avaliação de conformidade por meio de uma auditoria para todas as classes de dispositivos, exceto a Classe I. Isso significa que uma certificação ISO 13485 existente não é suficiente para que você possa reivindicar a conformidade com os requisitos do MDR QMS.
Transição da ISO 13485 para a MDR 2017/745: um guia
A transição da ISO 13485 para a MDR 2017/745 pode parecer complexa, mas, com a abordagem correta, pode ser um processo tranquilo. A primeira etapa é entender as diferenças entre as duas normas e identificar os requisitos adicionais impostos pela MDR 2017/745.
Em seguida, faça uma análise de lacunas para identificar as áreas em que o seu SGQ atual não atende aos requisitos da MDR 2017/745. Isso envolverá a revisão de procedimentos, documentação e práticas relacionadas a todos os estágios do ciclo de vida do dispositivo médico.
Após a análise das lacunas, desenvolva e implemente um plano para abordar as lacunas identificadas. Isso pode envolver a atualização de procedimentos, o aprimoramento da documentação ou o fortalecimento das atividades de vigilância pós-comercialização. Prepare-se para que esse seja um processo demorado e que exija o envolvimento de várias partes interessadas.
Por fim, certifique-se de que todas as alterações sejam comunicadas de forma eficaz em toda a organização e que seja fornecido o treinamento adequado. Isso garantirá que todos entendam os novos requisitos e possam contribuir para manter a conformidade.
Navegando pelo MDR 2017/745 e pela ISO 13485 para conformidade com o gerenciamento de qualidade
Navegar pelo mundo das regulamentações de dispositivos médicos, especialmente a MDR 2017/745 e a ISO 13485, pode ser complexo.
No entanto, a compreensão dessas normas e de suas diferenças pode ajudar os fabricantes a desenvolver sistemas robustos de gerenciamento de qualidade e a manter a conformidade.
Mesmo que a ISO 13485 não seja obrigatória, conforme demonstrado nos capítulos anteriores, ela é uma base sólida que facilitará significativamente o processo de implementação. Além disso, o forte foco na conformidade do produto, que precisa ser refletido no sistema de gerenciamento de qualidade, requer atenção especial.
Lembre-se, o objetivo do MDR 2017/745 e da ISO 13485 é garantir a segurança e a eficácia dos dispositivos médicos. Ao se concentrarem nesse objetivo comum, os fabricantes podem navegar com eficiência por essas regulamentações e ter sucesso no mercado global.
A transição de um sistema para outro, ou o gerenciamento da conformidade com ambos, pode ser um empreendimento significativo. No entanto, com planejamento cuidadoso, comunicação clara e compromisso contínuo, é possível atingir essa meta.
Se você precisar de suporte para entender a MDR 2017/745, a ISO 13485 ou para navegar na transição entre essas estruturas regulatórias, não hesite: Fale com nossos especialistas!


