Dans l’industrie des dispositifs médicaux, la gestion de la qualité et la conformité réglementaire sont d’une importance capitale. Deux réglementations importantes sont le règlement de l’Union européenne sur les dispositifs médicaux (MDR 2017/745) et la norme ISO 13485 de l’Organisation internationale de normalisation. Il est essentiel pour les fabricants de dispositifs médicaux de comprendre ces réglementations, car elles définissent les exigences d’un système de gestion de la qualité (SGQ) solide.
Le MDR 2017/745, mis en œuvre par l’Union européenne, est un règlement concernant la fabrication et la distribution des dispositifs médicaux sur le marché de l’UE. Il met l’accent sur la nécessité d’une plus grande transparence, d’une amélioration de la sécurité et d’une traçabilité accrue des dispositifs. D’autre part, la norme ISO 13485 prescrit les exigences d’un SMQ complet pour les dispositifs médicaux et bénéficie d’une reconnaissance internationale.
Pour garantir la conformité et la durabilité, les fabricants de dispositifs médicaux doivent se familiariser à la fois avec le MDR 2017/745 et la norme ISO 13485. Cette compréhension renforcera leur capacité à répondre aux exigences strictes des deux organismes de réglementation, ce qui leur donnera un avantage concurrentiel sur le marché mondial.
Comprendre l’ISO 13485 : Système de gestion de la qualité pour les dispositifs médicaux
La norme ISO 13485 définit les exigences relatives à l’élaboration d’un système de gestion de la qualité spécifique aux dispositifs médicaux et jouit d’une reconnaissance mondiale. L’objectif principal de cette norme est de faciliter l’harmonisation des exigences en matière de gestion de la qualité des dispositifs médicaux. La norme ISO 13485 met davantage l’accent sur la gestion des risques et les processus de prise de décision fondés sur les risques, ce qui la rend très différente des autres normes de SMQ.
Un système de gestion de la qualité ISO 13485 efficace garantit que les dispositifs médicaux répondent aux attentes des clients et sont conformes aux exigences réglementaires applicables. Cette norme met l’accent sur la maîtrise des processus, la satisfaction des clients, la traçabilité des produits et les systèmes de rappel, ainsi que sur le maintien de systèmes efficaces d’actions correctives et préventives.
L’adhésion à la norme ISO 13485 témoigne d’un engagement en faveur de la sécurité et de la qualité des dispositifs médicaux, ce qui peut renforcer la réputation d’une entreprise sur le marché. Cependant, comprendre les exigences de la norme ISO 13485 et mettre en œuvre un SMQ basé sur cette norme peut s’avérer une tâche difficile. Cela implique de développer des procédures qui garantissent des processus cohérents et conformes, ce qui demande beaucoup de temps et d’efforts.
Principales différences entre le MDR 2017/745 et la norme ISO 13485
Si le MDR 2017/745 et la norme ISO 13485 visent tous deux à garantir la sécurité et l’efficacité des dispositifs médicaux, il existe plusieurs différences essentielles. Comprendre ces différences peut aider les fabricants à naviguer plus efficacement dans ces exigences.
Contrairement à la norme ISO 13485, le MDR 2017/745 est un acte législatif, ce qui signifie qu’il est juridiquement contraignant. La norme ISO 13485 se concentre sur l’efficacité d’un système de gestion de la qualité. Il contient les processus et procédures requis et aucune exigence spécifique au produit. Le MDR 2017/745 se concentre davantage sur le dispositif médical lui-même avec des exigences détaillées, y compris sa conception, son développement, sa production et sa surveillance après la mise sur le marché.
Mais le règlement MDR prévoit-il des exigences supplémentaires en matière de SMQ ?
Oui, il y a quelques ajouts. Vous trouverez ci-dessous un aperçu des principaux thèmes à aborder dans votre système de gestion de la qualité.
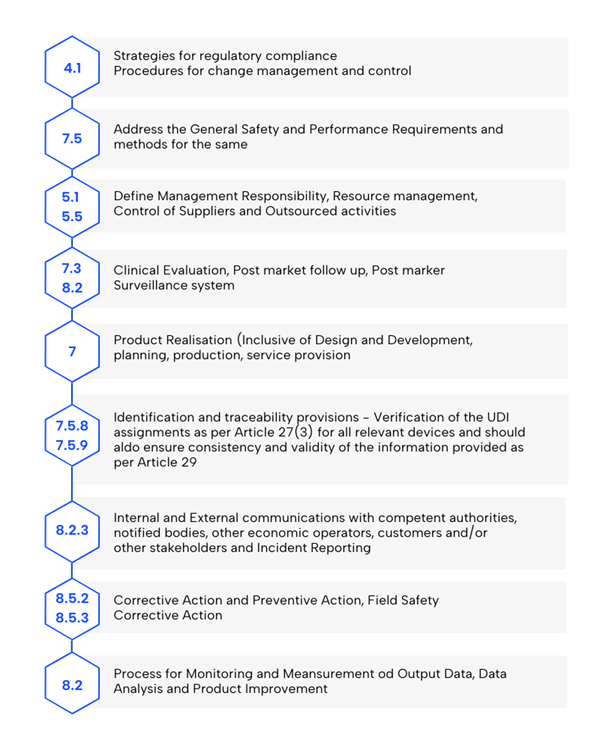
- Suivi et analyse des données: Établir des processus pour mesurer les résultats, analyser les données et améliorer les produits, en mettant l’accent sur les méthodes statistiques.
- Stratégie de conformité réglementaire: Doit inclure des procédures d’évaluation de la conformité et la gestion du changement.
- Exigences générales de sécurité et de performance (GSPR): Examinez les exigences de l’annexe I et les méthodes pour les respecter, en veillant à la conformité avec le MDR.
- Réalisation du produit: Comprend la planification, la conception, le développement, la production et la prestation de services, les RGPP faisant partie des exigences de conception.
- Personne responsable de la conformité réglementaire: Définir le rôle de la personne responsable de la conformité réglementaire conformément à l’article 15 du RIM.
- Gestion des ressources: Mettre en œuvre des méthodes de sélection et de contrôle des fournisseurs et des sous-traitants.
- Gestion des risques: Établir des processus conformément à la section 3, annexe I du MDR, la norme EN ISO 14971 comblant cette lacune réglementaire.
- Évaluation clinique et suivi clinique après commercialisation (PMCF): Inclure les données conformément à l’article 61 et à l’annexe XIV.
- Vérification de l’UDI: Assurer l’attribution correcte des dispositifs concernés et valider la cohérence des données (article 27, paragraphe 3, et article 29).
- Surveillance après la mise sur le marché: Établir et maintenir un système tel que requis par l’article 83.
- Protocoles de communication: Définir les interactions avec les autorités, les organismes notifiés, les opérateurs économiques, les clients et les parties prenantes.
- Rapport sur les événements graves et les actions correctives: Inclure des méthodologies pour la vigilance et les actions correctives en matière de sécurité sur le terrain (article 87).
- Gestion des actions correctives: Mettre en œuvre et vérifier les actions correctives afin d’en garantir l’efficacité.
Quelles sont les parties de la norme ISO 1385 les plus concernées ?
Une autre différence importante est que le MDR 2017/745 s’applique exclusivement au marché européen, alors que la norme ISO 13485 est reconnue au niveau mondial.
Cela signifie qu’un SMQ certifié ISO 13485 peut être utilisé presque partout dans le monde, alors que la conformité au MDR 2017/745 est nécessaire pour accéder au marché européen.
Malgré ces différences, le MDR 2017/745 et la norme ISO 13485 ont un objectif commun : garantir la sécurité et la performance des dispositifs médicaux. Ils soulignent tous deux l’importance d’un SMQ solide, de la gestion des risques et de l’amélioration continue.
Comment un système de gestion de la qualité sera-t-il évalué dans le cadre du règlement MDR de l’UE ?
Le système de gestion de la qualité d’un fabricant sera évalué dans le cadre des procédures d’évaluation de la conformité des annexes IX à XI du RMD de l’UE. Un organisme notifié effectuera une évaluation de la conformité par le biais d’un audit pour toutes les classes de dispositifs, à l’exception de la classe I. Cela signifie qu’une certification ISO 13485 existante n’est pas suffisante pour prétendre au respect des exigences du MDR en matière de système de gestion de la qualité.
Passer de la norme ISO 13485 à la norme MDR 2017/745 : un guide
Le passage de la norme ISO 13485 au règlement MDR 2017/745 peut sembler complexe, mais avec la bonne approche, le processus peut se dérouler sans heurts. La première étape consiste à comprendre les différences entre les deux réglementations et à identifier les exigences supplémentaires posées par le MDR 2017/745.
Ensuite, procédez à une analyse des lacunes pour identifier les domaines dans lesquels votre SMQ actuel ne répond pas aux exigences du MDR 2017/745. Il s’agira d’examiner les procédures, la documentation et les pratiques liées à toutes les étapes du cycle de vie des dispositifs médicaux.
À la suite de l’analyse des lacunes, élaborez et mettez en œuvre un plan visant à combler les lacunes identifiées. Il peut s’agir de mettre à jour les procédures, d’améliorer la documentation ou de renforcer les activités de surveillance après la mise sur le marché. Préparez-vous à ce que ce processus prenne du temps et nécessite l’implication de diverses parties prenantes.
Enfin, veillez à ce que tous les changements soient communiqués efficacement à l’ensemble de l’organisation et à ce qu’une formation adéquate soit dispensée. Ainsi, tout le monde comprendra les nouvelles exigences et pourra contribuer au maintien de la conformité.
Naviguer entre MDR 2017/745 et ISO 13485 pour la conformité de la gestion de la qualité
Naviguer dans le monde des réglementations relatives aux dispositifs médicaux, en particulier le MDR 2017/745 et la norme ISO 13485, peut s’avérer complexe.
Cependant, la compréhension de ces réglementations et de leurs différences peut aider les fabricants à développer des systèmes de gestion de la qualité solides et à maintenir la conformité.
Même si la norme ISO 13485 n’est pas obligatoire, comme l’ont montré les chapitres précédents, elle constitue une base solide qui facilitera considérablement le processus de mise en œuvre. De même, l’accent mis sur la conformité des produits, qui doit se refléter dans le système de gestion de la qualité, doit faire l’objet d’une attention particulière.
N’oubliez pas que l’objectif du MDR 2017/745 et de la norme ISO 13485 est de garantir la sécurité et l’efficacité des dispositifs médicaux. En se concentrant sur cet objectif commun, les fabricants peuvent naviguer efficacement dans ces réglementations et réussir sur le marché mondial.
Le passage d’un système à l’autre, ou la gestion de la conformité avec les deux, peut être une entreprise importante. Cependant, avec une planification minutieuse, une communication claire et un engagement continu, c’est un objectif réalisable.
Si vous avez besoin d’aide pour comprendre le MDR 2017/745, l’ISO 13485 ou pour naviguer dans la transition entre ces cadres réglementaires, n’hésitez pas : Parlez-en à nos experts !


